Introduction
Turfgrasses have been an integral part in the landscaping industry, playing an important part in the urban and suburban environment. The turfgrass industry has a huge impact, both environmentally and economically (Haydu et al., 2006). Among the warm-season turfgrasses, the members of the genera Zoysia are one of the most resilient turfgrasses in terms of their abiotic stress tolerance (Huang et al., 2014). Zoysiagrasses are preferred as turfgrasses because they are salt tolerant, heat and cold tolerant and are aesthetically pleasing (Feng et al., 2019; Patton, 2010). High density genetic linkage mapping has been done in the three zoysiagrasses; Z. japonica, Z. matrella and Z. pacifica, revealing high introgression and hybridization rates (Tanaka et al., 2016). The acquisition of this huge sequencing resource has enabled researchers to find the molecular mechanisms involved in abiotic and biotic stress tolerance, and to further utilize this information to create better cultivars.
Climate change has initiated a new era of research to identify plants that can thrive in harsh abiotic stress conditions. Unfavorable environmental conditions cause the plants to employ intricate molecular networks involving transcription factors (TFs) that further regulate specific gene expressions, helping the plant cope with stress. One such family of TFs is the APETALA2/ethylene response factor (AP2/ERF) family, characterized by a AP2 DNA binding domain. Their functions range from controlling the developmental and physiological aspects of plant growth to providing defense response in case of abiotic or biotic stress conditions (Shoji and Yuan, 2021). The family can further be divided into AP2, ERF, related to ABI3/VP (RAV), based on the number of AP2 domains and the presence of other DNA binding domains. Members of the subfamily ERF have a single AP2 domain, that specifically binds to GCC-boxes, and are documented to be involved in jasmonate and ethylene signaling and abiotic stress response (Feng et al., 2020).
In Arabidopsis, ERF1 has been reported to be highly induced by salt and drought stress (Cheng et al., 2013). In Z. japonica, ERF genes (ZjERF1, ZjERF2) have been isolated and are reported to be induced by ethylene, methyl jasmonate and high salinity conditions (Teng et al., 2019). Studies have shown that Z. matrella and Z. pacifica can regulate tissue salt levels more efficiently than Z. japonica (Marcum et al., 1998; Yamamoto et al., 2016). One of the key features in the salt stress tolerance in zoysiagrasses is the presence of salt glands on the leaf surface which help in the secretion of Na+ (Marcum et al., 1998). So far, no information regarding the ERF genes in Z. matrella and Z. pacifica has been published to deduce the underlying genetic mechanism of abiotic and biotic stress. The objective of this study is to (i) predict ERF genes in Z. matrella and Z. pacifica, (ii) Structural and functional characterization of the predicted ERF genes using phylogenetic analysis and protein structure prediction (iii) to identify the probable effects of the variations in the ERF gene among the three zoysiagrasses on the abiotic stress tolerance properties of the zoysiagrasses.
Materials and methods
Sequence data retrieval and identification of ERF genes in zoysiagrass
Zoysia japonica (Zj), Zoysia matrella (Zm), and Zoysia pacifica (Zp) genome sequences were downloaded from ‘Zoysia Genome Database’ (http://zoysia.kazusa.or.jp/). The full coding sequence and protein sequence of Z. japonica ERF1 and ERF2 was obtained from NCBI (https://www.ncbi.nlm.nih.gov/). This sequence was used as a query for a BLAST (Basic Local Alignment Search Tool) search against Z. pacifica and Z. matrella genome with the following parameters: expected values ≤1E-5 and more than 80 percent coverage. SNPs and in/dels were identified using SNP-sites (Page et al., 2016). All the BLAST hits were retrieved, and a conserved domain search was performed using NCBI conserved domain search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Multiple alignments of the putative ERF genes were done using ClustalW (https://www.genome.jp/tools-bin/clustalw) (Thompson et al., 1994) with the default settings. Phylogenetic analysis of the ERF transcription factor genes was done using the MEGA X (https://www.megasoftware.net/) maximum likelihood method (Kumar et al., 2018).
Identification of ERF upstream regulatory region
The upstream regulatory regions of the Zoysia ERF genes were identified by extracting a 1.7 kb sequence upstream of the transcription start site. The putative promoter and TATA box sites were predicted using Transcription Start Sites Plant (TSSP) (Shahmuradov et al., 2017). Further, PlantRegMap (Tian et al., 2020) was used to identify transcription factor binding sites near the predicted ERF1/2 upstream regulatory regions. Gene Ontology (GO) enrichment analysis was done to identify TF binding sites significantly related to abiotic and biotic stress response signaling (Alexa and Rahnenführer, 2009).
Protein structure prediction and evolutionary conservation analysis
The amino acid sequence of the predicted Zoysia ERF genes was used to predict the putative tertiary structure of the proteins. ProtParam tool from Expasy was used to compute the physical and chemical parameters if the predicted proteins (Gasteiger et al., 2005). Protein structure prediction was done using Protein Homology/analogY Recognition Engine 2 (PHYRE2) (Kelley et al., 2015) and Chimera 1.15 was used for protein structure visualization. The ConSurf server (http://consurf.tau.ac.il/) estimated the conservation pattern of each amino acid in the identified ERF to measure the degree of conservation at each aligned site. The ConSurf scores vary from 1 to 9, with 1 being for fast-evolving (variable) sites. Active ligand binding sites of the proteins were detected using fPocket2 (http://fpocket.sourceforge.net/).
Results and Discussion
Identification of Zoysia ERF transcription factors
The identification of the ERF transcription factors in Z. matrella and Z. pacifica was done using Z. japonica ERF1 and ERF2 full length coding sequences that have been already deposited to the GenBank; accessions MH294481.1 and MH479420.1 respectively. This sequence was used a query for a BLAST search against the Z. matrella and Z. pacifica genomes. In Z. matrella, two sequence hits were obtained for both ERF1 and ERF2 TFs, which were denoted as ZmERF1a/b and ZmERF2a/b respectively (Table 1). The ERF1 genes had no exons, whereas ERF2 genes in all the zoysiagrasses displayed two exons (Fig. 1). Many studies have reported that the number of exons in the ERF gene vary from one to three or more (Ma et al., 2015). The open reading frame in ZmERF1a was 630 bp long encoding 209 amino acids, identical to reports in the ZjERF1 (Teng et al., 2019) . However, ZmERF1b and ZpERF1 had slightly shorter open reading frame of 618bp encoding 205 amino acids. Analysis of SNPs in the predicted ERF1 TFs revealed that both ZmERF1a and ZpERF1 had 6 SNPs and a 12bp deletion with respect to ZjERF1 (Table 2A). Additionally, ZmERF1b had a single nucleotide polymorphism at position 337 (Table 2A). The SNP analysis in the ERF2 genes with respect to the ZjERF1, in the three zoysiagrasses revealed 1, 3 and 5 SNPs in ZmERF2a, ZMERf2b and ZpERF2 respectively (Table 2B). Conserved domain analysis showed that all the ERF genes identified in the zoysiagrasses consisted of one AP2 domain, which is indicative of a typical ERF family transcription factor (Phukan et al., 2017).
|
Table 1. Zoysia ERF1 and ERF2 predicted genes and their sequence information. 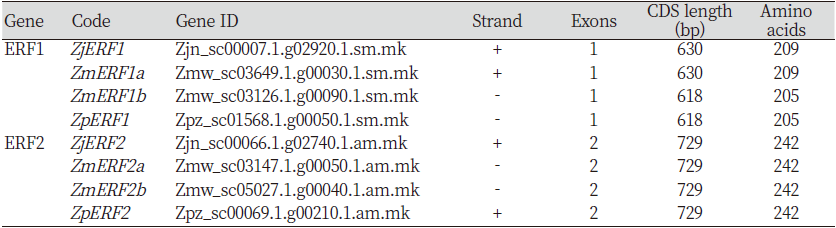
|
|
ERF: Ethylene responding factor; CDS: Coding sequence; Zj: Z. japonica; Zm: Z. matrella; Zp: Z. pacifica. |

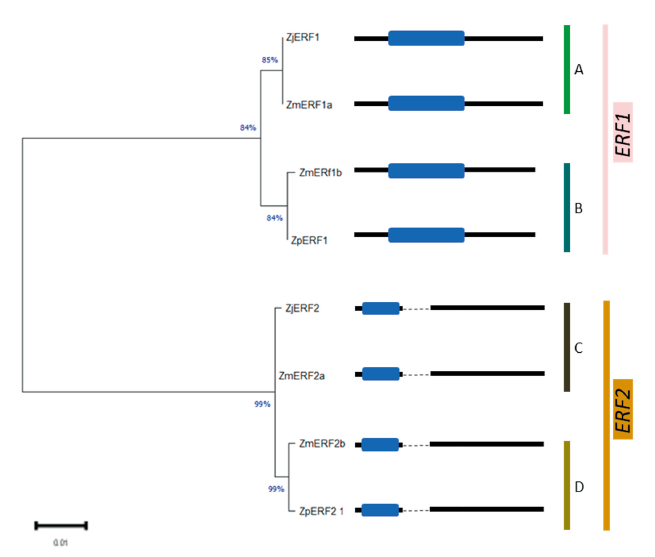
Fig. 1. Phylogenetic relationship of ERF transcription factor genes among the three zoysiagrasses using maximum likelihood method with 500 bootstrap replications. The blue boxes represent AP2 domain, black lines represent exons and dotted lines represent introns. ERF: Ethylene Response Factor; AP2: APETALA2; Zj: Zoysia japonica; Zm: Zoysia matrella; Zp: Zoysia pacifica.
Analysis of the upstream regulatory region of the ERF Transcription factor genes
Sequences 1.7 kb upstream of the transcription start site were used to identify putative promoter regions. All ERF1 and ERF2 genes constituted at least one promoter accompanied by a TATA box. The ERF1 predicted promoter region was seen to be highly conserved, whereas the ERF2 promoter region had high variability. In the case of ERF1 in all the three zoysiagrasses constituted one predicted promoter and TATA box site at the same position, with the exception of ZjERF1, having an additional predicted promoter at position -573 and an accompanying TATA box at -544. Each zoysiagrass consisted of two highly conserved promoter regions in the case of ERF2 gene.
Using PlantTFDB (http://plantregmap.gao-lab.org/binding_site_prediction_result.php), top 50 TF binding sites in the 1.7 kb upstream regulatory region (URR) of the transcription start site (TSS) were identified. The results were filtered by removing the duplicates, and only values above q value <0.05 were selected to eliminate false positives. There were more TF binding sites in the URR of the ERF2 gene than in ERF1. Common TF binding sites in both the ERF genes among the three zoysiagrasses included Dof, MIKC_MADS, Trihelix.
GO enrichment analysis was conducted to identify the TF binding sites related to the documented ERF TFs functions. The ERF1 TF binding sites in the URR revealed many motifs, that were related to responses to plant development and flower regulation, responses to plant hormones and biotic and abiotic stresses. The motif jn_sc00045.1.g02180.1.sm.mk in ZjERF1, belonging to Dof family, engages in a wide range of responses like response to drought salinity plant hormones, bacteria, and fungi. The motifs Zjn_sc00007.1.g03660.1.sm.mk and Zmw_sc01431.1.g00150.1 belonging to ERF family were related to response to cold and heat stress, and response to salicylic acid and jasmonic acid. The TF binding sites in Z. pacifica ERF1 were related to floral organ identity, flower development fruit development. Motifs related to response to abiotic and biotic stresses were absent in the ZpERF1 URR (Table S1).
Transcription factor binding sites in the ERF2 URR of the four genes were related to the WRKY, MIKC_MADS, HD-ZIP and MYB_related TF families (Table S2). The motifs Zjn_sc00024.1.g02600.1.sm.mkhc and Zjn_sc00075.1.g00940.1.am.mkhc in the ZjERF2 URR belong to the WRKY family and are involved jasmonic acid mediated pathway, response to salicylic acid and defense response to fungal and bacterial attack. The ZmERF2a,b URRs also consisted of motifs belonging to the WRKY TF family, which respond to salicylic acid, chitin, and fungal and bacterial attack. The motifs belonging to HD-ZIP family in ZmERF2a,b (Zmw_sc00963.1.g00120.1, Zmw_sc00963.1.g00120.1), may have a role in salt stress or osmotic stress response. In Z. pacifica ERF2, motifs belonging to the WRKY family (Zpz_sc00228.1.g00240.1.sm.mk, Zpz_sc02744.1.g00050.1.sm.mkhc, Zpz_sc00990.1.g00040.1.sm.mkhc) may respond to jasmonic acid stimulus, and fungal and bacterial attacks. The motif Zpz_sc00747.1.g00070.1.sm.mk, belonging to WRKY family may be involved in the negative regulation of gibberellic acid pathway.
Most of the TF binding sites had a redundant function, specifically in the two copies of the Z. matrella ERF1 and ERF2 transcription factors. Such redundant TF clusters may indicate gene regulatory hotspots (Dergilev et al., 2022).
Phylogenetic relation of the ERF gene in the three Zoysiagrasses
To determine the evolutionary relationship among the ERF genes, an unrooted phylogenetic tree based on the maximum likelihood method was constructed using MEGA X based on the multiple sequence alignment of the 8 ERF transcription factor genes. The phylogenetic tree showed that ERF1 and ERF2 were distinctly separated into two major groups, which were further divided into two subgroups. ZmERF1a was found to be closely related to ZjERF1 (subgroup A), whereas ZmERF1b and ZpERF1 were grouped together (subgroup B) (Fig. 1). In the ERF2 group, further two subgroups were formed with ZjERF2 and ZmERF2a forming subgroup C and ZmERF2b and ZpERF2 grouped together in subgroup D (Fig. 1).
Evolutionary-based conservation analysis of the predicted ERF1/2 proteins
The ERF1/2 amino acid sequences obtained from the three zoysiagrasses were aligned using ClustalW. The amino acids in the AP2 binding domain of the ERF1 protein were marked as highly conserved by ConSurf. Within the AP2 conserved domain, one amino acid substitution was observed at position 64, wherein the Alanine in ZjERF1 and ZmERF1 is substituted with Threonine in ZmERF1b and ZpERF1 (Fig. 2A). This missense mutation is a result of a single base substitution in the codon: GCC (producing alanine) to ACC (producing threonine). Both the amino acids were seen to be categorized as evolutionary conserved in the ConSurf analysis, implying that the amino acid changes at the position 64 may affect the structure or function of the protein drastically. Outside the AP2 conserved domain, another amino acid substitution was seen at position 113, where Aspartic acid was substituted by Histidine in ZmERF1b. The amino acids at the position 113 were categorized as highly variable by ConSurf analysis, leading to a conclusion that they may have little to no effect the properties of the ERF1 protein.
Among the ERF2 proteins, the amino acids in the AP2 domain were highly conserved evolutionary, and no amino acid substitutions were observed within this domain. Outside the AP2 domain, two amino acid substitutions were seen; the first one at position 20, where Arginine in ZjERF2 and ZmERF2a was substituted to Histidine in ZmERF2b (Fig. 2b). The second substitution was seen at position 224, where ZjERF2 has Threonine and the other three zoysiagrass ERF2 show a Methionine. Both positions were highly variable in the evolutionary conservation analysis and any substitutions at this position may have negligible effects on the protein structure and function.
Protein structure prediction and the analysis of the AP2 binding domain
The physical and chemical properties of the predicted genes were analyzed using the ProtParam tool on Expasy (https://web.expasy.org/protparam) Gasteiger et al., 2005(). The ZjERF1 and ZmERF2a proteins had a comparatively similar molecular mass but a slightly higher pH than the ZmERF1b and ZpERF1 proteins (Fig. 3). In the four zoysiagrass ERF2 proteins the chemical and physical properties were slightly variable (Fig. 4). The prediction of the three-dimensional structure of the predicted Zoysia ERF1 and ERF2 genes was done to see the probable effect of the amino acid substitutions. Phyre2 protein structure prediction relies on the evolutionary and statistical comparison of the query amino acid sequence against an ever-growing database of proteins with known structures Kelley et al., 2015().
The ERF1 and ERF2 protein structures were highly disordered (60-62%) in all the zoysiagrasses. Intrinsically disordered proteins (IDPs) make up almost 30-50% of the eukaryotic proteins. These proteins are biologically active, but are unable to adopt persistent tertiary structures, leading to dynamic conformational ensembles (Salladini et al., 2020). The AP2/ERF family domain binding sites are flanked with intrinsically disordered residues, lacking secondary structures. ZjERF1, ZmERF1a and ZpERF1 protein constituted of 24% of the structure forming alpha helices and the 9% forming beta sheets (Fig. 3). ZmERF1b had the lowest structural disorder (57%) among the other zoysia ERF1 proteins, with a structure consisting of 24% alpha helices and 9% beta sheets (Fig. 3). ZjERF2 and ZmERF2a protein structures constituted 30% alpha helices and 7% beta sheets, whereas ZmERF2b and ZpERF2 protein structures constituted 28% alpha helices and 5% beta sheets (Fig. 4).
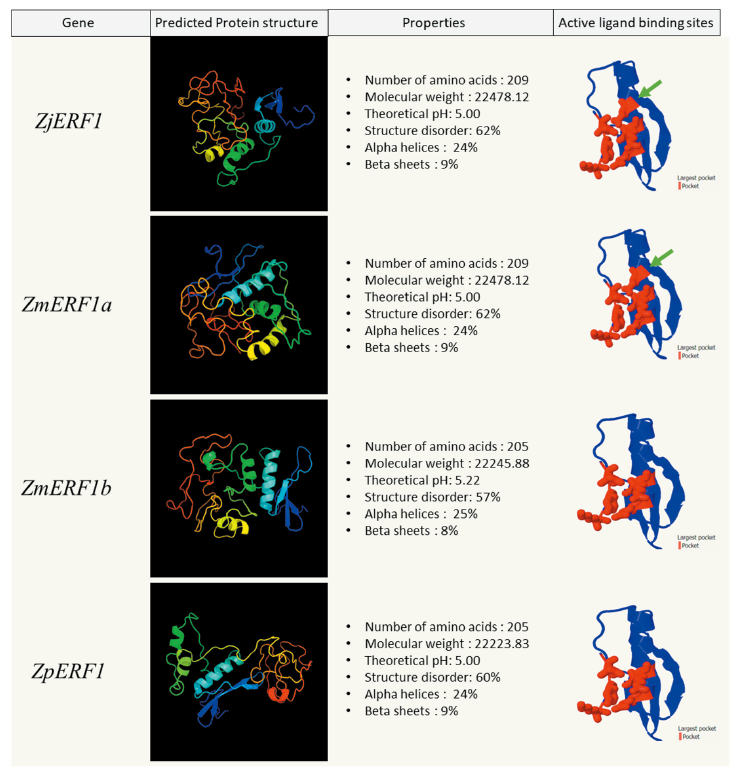
Fig. 3. Zoysia ERF1 proteins in the physical, chemical, and structural analysis. For each ERF1 protein, the protein structure was predicted using PHYRE2 (image colored by rainbow N to C terminus). Largest active ligand binding sites (in red) were predicted using fPocket2. Green arrows denote an additional active binding site in ZjERF1 and ZmERF2a. ERF: Ethylene Response Factor; Zj: Zoysia japonica; Zm: Zoysia matrella; Zp: Zoysia pacifica.
Protein homology search of the ERF1 and ERF2 predicted sequences yielded a significant hit to the GCC-box binding domain, which is a core function of the ERF transcription factors (Zarei et al., 2011). Protein pocket detection can reveal the functional & ligand binding sites in a protein (Stank et al., 2016). The detection of active ligand binding sites in the GCC-box binding domain of the ERF proteins was done using fPocket2. The active pocket of ZjERF1 and ZmERF1a were identical whereas the pockets of ZmERF1b and ZpERF1 were identical to each other (Fig. 3). The difference among these two groups was seen as a result of an amino acid substitution at position 64 (Alanine → Threonine) in the AP2/ERF domain (Fig. 2). The active binding site in the GCC-box-domain of all the four ERF2 TFs was identical, owing to the fact that there were no amino acid substitutions seen in the conserved AP2/ERF domain (Fig. 4).
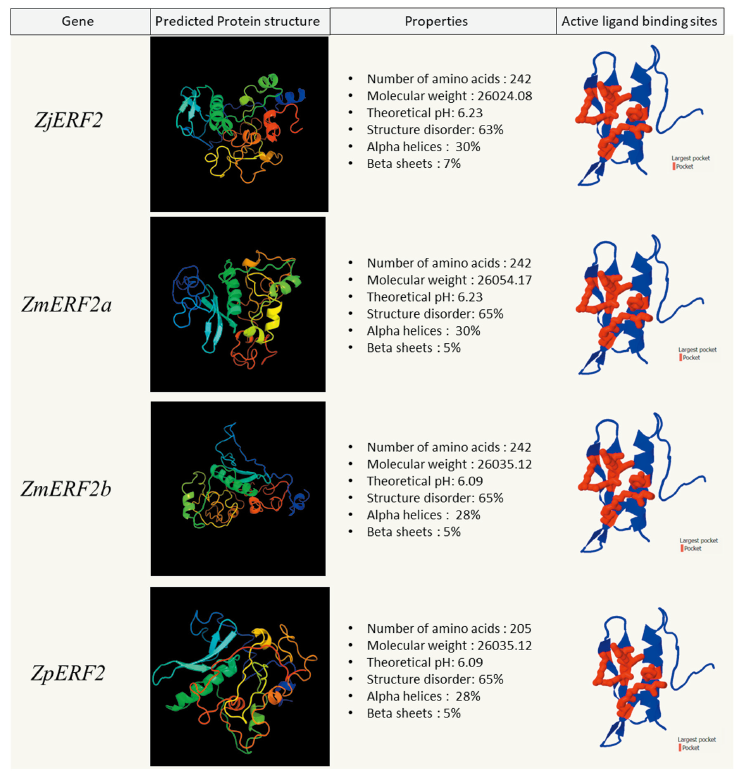
Fig. 4. Zoysia ERF2 proteins physical, chemical, and structural analysis. For each ERF2 protein, the protein structure was predicted using PHYRE2 (image colored by rainbow N to C terminus) . Largest active ligand binding sites (in red) were predicted using fPocket2. ERF: Ethylene Response Factor; Zj: Zoysia japonica; Zm: Zoysia matrella; Zp: Zoysia pacifica.
Conclusion
In the wake of climate change, zoysiagrasses have recently come under the spotlight to explore their genomes and identify genes that confer better survival in adverse environmental conditions. Members of the zoysiagrasses like Z. japonica have been widely studied for its cold and drought tolerance (Cohen et al., 2019; Kaur et al., 2022). Studies on the salt tolerance mechanism in the zoysiagrasses revealed that Z. matrella and Z. pacifica have a higher salt tolerance capability than Z. japonica (Loch et al., 2005; Marcum et al., 1998; Sugiura and Takahashi, 2021). The varying degrees of salt tolerance among the zoysiagrasses was attributed to leaching (Loch et al., 2005) , but the key genetic players behind the salt tolerance capability have been poorly studied. Z. japonica , Z. matrella and Z. pacifica show poor drought tolerance capabilities when compared to Bermudagrass (Loch et al., 2017). ERF gene family is an excellent candidate to study the differential regulation of genetic mechanisms that confer biotic and abiotic stress resistance in plants (Owji et al., 2017).
The present study aimed to predict the ERF1 and ERF2 TF genes in Z. matrella and Z. pacifica and to compare the probable effects of the variation in the gene structure among the Zoysia grasses. Phylogenetic analysis divided the eight ERF TFs to two groups, which was consistent with the previous studies where the basis of classification was the presence of introns and conserved motifs (Ma et al., 2015). The phylogenetic grouping was further validated by the protein structure analysis, wherein the ERF TF genes belonging to the same subgroups has similar structure and chemical properties. The ERF TFs are known to be involved in hormonal, abiotic stress and biotic stress signaling pathways (Shoji and Yuan, 2021). The TF binding motifs in the upstream regulatory region of each ERF gene had at least one motif related to these signaling pathways, with the exception of ZpERF1 having no significant motifs related to abiotic or biotic stress signaling. The effect of the single amino acid substitution at the 64th amino acid between the sub-group A (ZjERF1, ZmERF1a) and sub-group B (ZmERF1b, ZpERF1) was seen in the analysis of the GCC-box-binding domain, wherein the presence of Alanine in sub-group A conferred an additional active site. The experimental validation of the effect of Ala64 to Thr64 substitution can provide some clues in understanding the variation in the abiotic stress response among the three zoysiagrasses. The probable effect of the TF binding motifs in the upstream regulatory regions of the ERF transcription start site may also be studied further to identify their roles in enhancing stress signaling mechanisms for in Zoysia. The information provided in this study aims to add to the otherwise scarce resource of the zoysiagrass research pool.
Acknowledgment
This research was funded by the New Breeding Technologies Development Program (No. PJ016547), Rural Development Administration, and the Basic Science Research Program (NRF-2020R1A2C1015119) through the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT, Republic of Korea.

