Introduction
Zoysiagrass is a representative turfgrass species distributed from temperate regions in Northeast Asia, including Korea, to tropical areas in Southeast Asia, such as the Philippines and Thailand (Engelke et al., 1983). It is used in golf courses, lawns, sports turf, roadsides, restoration sites, and other areas. Current research on zoysiagrass has primarily focused on habitat surveys, sample collection, growth assessments, artificial hybridization, and mutation breeding to develop new cultivars (Bae et al., 2010a; Bae et al., 2010b; Yang et al., 2023). Additionally, research on gene overexpression, genetic transformation, and related topics is being conducted continuously and there is a need to explore genes that can be used for the improvement of zoysiagrass (Kang et al., 2016; Kim et al., 2004; Ng et al., 2023; Zhang et al., 2007; Zuo et al., 2019).
Ascorbate peroxidase (APX) was initially predicted through the discovery of an ascorbate-dependent system involved in H2O2 removal (Groden and Beck, 1979), and its presence was confirmed with the identification of soluble APX (Kelly and Latzko, 1979). Later, it was identified in spinach, enabling the immunochemical verification of its presence across various plant species (Saji et al., 1990). Finally, cytosolic APX (cAPX) was purified from pea (Pisum sativum), facilitating a deeper understanding of its molecular properties and physiological roles (Mittler and Zilinskas, 1991).
APX primarily removes reactive oxygen species (ROS) using ascorbate, but under abiotic stresses like heat, high light, cold, and salt, its activity increases to manage elevated ROS levels and helps the plant to adapt. In rice, OsAPX2 gene expression was upregulated 2.5-fold in response to salt stress (Teixeira et al., 2006). Overexpression of APX genes in transgenic plants led to an improved tolerance to salinity and oxidative stress (Lu et al., 2007; Wang et al., 2005; Zhang et al., 2014).
APX is a potential target of tyrosine nitration, a post-translational modification that can impact its function, and its activity can be regulated by nitric oxide (NO) through various mechanisms, supporting the enzyme’s response to environmental stress (Keyster et al., 2011; Lin et al., 2011; Tanou et al., 2012). The APX gene family has been studied through in silico methods, in species like Arabidopsis thaliana and Oryza sativa (Jespersen et al., 1997; Teixeira et al., 2004). APX isoforms are distributed across at least four cellular compartments: stromal APX (sAPX) and thylakoid membrane-bound APX (tAPX) in chloroplasts, microbody membranebound APX (mAPX) in organelles like glyoxysomes and peroxisomes, and cAPX in the cytoplasm (Shigeoka et al., 2002). In the present study, we focused on predicting cAPX among the APX isoforms in four species of zoysiagrasses using bioinformatics tools. Three APX genes were predicted in each species, with an exception of four in Z. matrella. We report the analysis of the gene structure, the distribution of the transcription start site (TSS) and transcription factor (TF) binding sites, the phylogenetic relationship of the fourteen APX genes and their protein structure prediction. This study aims to understand the effect of genetic variation in different APXs in the four species of zoysiagrasses using comparative genomics approaches, focusing on predicting how single amino-acid polymorphisms (SAPs) or in/dels lead to protein structural variations and influence the catalytic activity, stress-response efficiency, or substrate specificity of APXs.
Materials and methods
Identification and sequence data retrieval of Zoysia ascorbate peroxidase (APX) genes
The full coding sequence and genomic DNA sequence of the APX gene from A. thaliana (AtAPX) and O. sativa (OsAPX) was obtained from the National Center for Biotechnology Information (NCBI) database (Wheeler, 2007). The genomic sequences of Z. japonica (Zj), Z. matrella (Zm), and Z. pacifica (Zp) were downloaded from the Zoysia Genome Database (Tanaka et al., 2016). The genomic sequence of Z. sinica (Zs) was obtained from the whole genome sequencing data (unpublished data). To identify APX genes in Z. japonica, Z. pacifica, Z. matrella, and Z. sinica, a Basic Local Alignment Search Tool (BLAST) search using the AtAPX1/AtAPX2 and OsAPX1/OsAPX2 sequence as queries was performed using the following parameters: expected values ≤ 1e-100 and more than 80% coverage (Altschul et al., 1990). After retrieving all of the BLAST results, the NCBI Conserved Domain database was used to conduct a search for conserved domains (Marchler-Bauer et al., 2017; Wang et al., 2023). The intron-exon structures of the predicted APX genes were manually constructed by aligning the coding sequence (CDS) with their corresponding genomic sequences. The phylogenetic tree was constructed with Molecular Evolutionary Genetics Analysis (MEGA) 11, using maximum likelihood method and JTT matrix-based model (Jones et al., 1992) with 500 bootstrap replications (Tamura et al., 2021).
Identification of Zoysia APX upstream and downstream regulatory region
A 1.5 kb region upstream of the CDS was extracted to identify the TSS and TATA box. Transcription Start Sites Plant (TSSPlant) was used to predict potential TSS and TATA box sites (Shahmuradov et al., 2017). For TSS with a TATA box, data above the threshold of 1.52 were used, and for TSS without a TATA box, data above the threshold of -0.04 were used. The TSS closest to the CDS was designated as +1, and an 800 bp upstream sequence was analyzed. Transcription factor binding sites near TSS were predicted using Plant Transcription Factor Database (Jin et al., 2017) and Plant Regulation Data and Analysis Platform (Tian et al., 2020), with a threshold of p-value ≤ 1e-4 and analysis restricted to the plus strand.
Protein structure prediction
The physical and chemical parameters of the predicted APX proteins in the four species of zoysiagrasses were calculated using ProtParam (Walker, 2005). The tertiary structures of the APX proteins were predicted using SWISS-MODEL (Bertoni et al., 2017; Bienert et al., 2017; Guex et al., 2009; Studer et al., 2020; Waterhouse et al., 2018), with templates that had a sequence identity ≥ 85%. Both tools are available on Expasy.
Results and Discussion
Identification and characterization of the APX genes in the four species of zoysiagrasses
In zoysiagrass, research on APX has been conducted, such as analyzing gene expression under stress conditions (Sun et al., 2019), but research on the gene itself is insufficient. Therefore, the cAPX genes, APX1 (NCBI gene ID: 837304, 4332474) and APX2 (NCBI gene ID: 820121, 4344397), from A. thaliana and O. sativa were used to predict the genes through BLAST. As a result, a total of 14 significant BLAST results were identified (Table 1). When comparing the BLAST results, the expected values aligned with A. thaliana ranged from 7e-161 to 3e-149, while those aligned with O. sativa ranged from 1e-175 to 6e-165. This indicates higher alignment reliability with O. sativa, as zoysiagrass is phylogenetically closer to the monocot O. sativa than the dicot A. thaliana.
Among the nine predicted APX1 group genes, six (ZjAPX1_1, ZmAPX1_1, ZmAPX1_2, ZpAPX1_1, ZsAPX1_1, ZsAPX1_2) had nine exons and eight introns, with the CDS length being 753 bp, commonly encoding 250 amino acids. ZjAPX1_2 had 11 exons and 10 introns, with a 933 bp CDS encoding 310 amino acids. ZmAPX1_3 and ZpAPX1_2 shared the same structure with 10 exons and 9 introns, but had slight differences in length, with CDS lengths of 768 bp and 777 bp, encoding 255 and 258 amino acids, respectively.
On the other hand, the five APX2 genes were all predicted to have seven exons and six introns. With an exception of ZmAPX2_2 and ZpAPX2, which had a CDS length of 891 bp encoding 296 amino acids, the other four genes were predicted to have a CDS length of 753 bp, encoding 250 amino acids (Table 1). In O. sativa, a monocot plant like zoysiagrass, two cAPX genes have been reported, both with nine exons and eight introns. OsAPX1 has a CDS length of 753 bp encoding 250 amino acids (Teixeira et al., 2004), consistent with most of the four predicted APX1s in zoysiagrass, suggesting that the APX1 gene has a conserved structure.
Conserved domain analysis of the predicted cAPX proteins revealed specific hits for the ascorbate peroxidase domain (cd00691) in the APX1 group. However, the APX2 group was predicted to belong to the larger plant peroxidase-like superfamily domain (cl00196). This is consistent with the findings in O. sativa, where OsAPX1 (NCBI gene ID: 4332474) contains the ascorbate peroxidase domain, and OsAPX2 (NCBI gene ID: 4344397) belongs to the plant peroxidase-like superfamily domain. Although ZsAPX1_1 was predicted to belong to the plant peroxidase-like superfamily domain by the NCBI Conserved Domain database, it was judged more appropriate to classify it as an ascorbate peroxidase domain based on phylogenetic relationship analysis and multiple sequence alignment (Fig. 1, 2).
Using the maximum likelihood method to analyze the phylogenetic relationships among the 14 predicted genes, two major groups were identified: APX1 (Group A) and APX2 (Group B). Each group was further divided into subgroups: Group A into four subgroups (a: ZpAPX1_1, ZsAPX1_2, ZmAPX1_1; b: ZjAPX1_1, ZmAPX1_2; c: ZjAPX1_2, ZmAPX1_3; d: ZpAPX1_2, ZsAPX1_1), and Group B into two subgroups (e: ZmAPX2_2, ZpAPX2; f: ZsAPX2, ZjAPX2, ZmAPX2_1) (Fig. 1). The phylogenetic analysis results also clearly show a division into APX1 and APX2, which is in agreement with the previously observed conserved domain analysis.
Table 1
List of APX genes predicted in the four species of zoysiagrass and their sequence information.
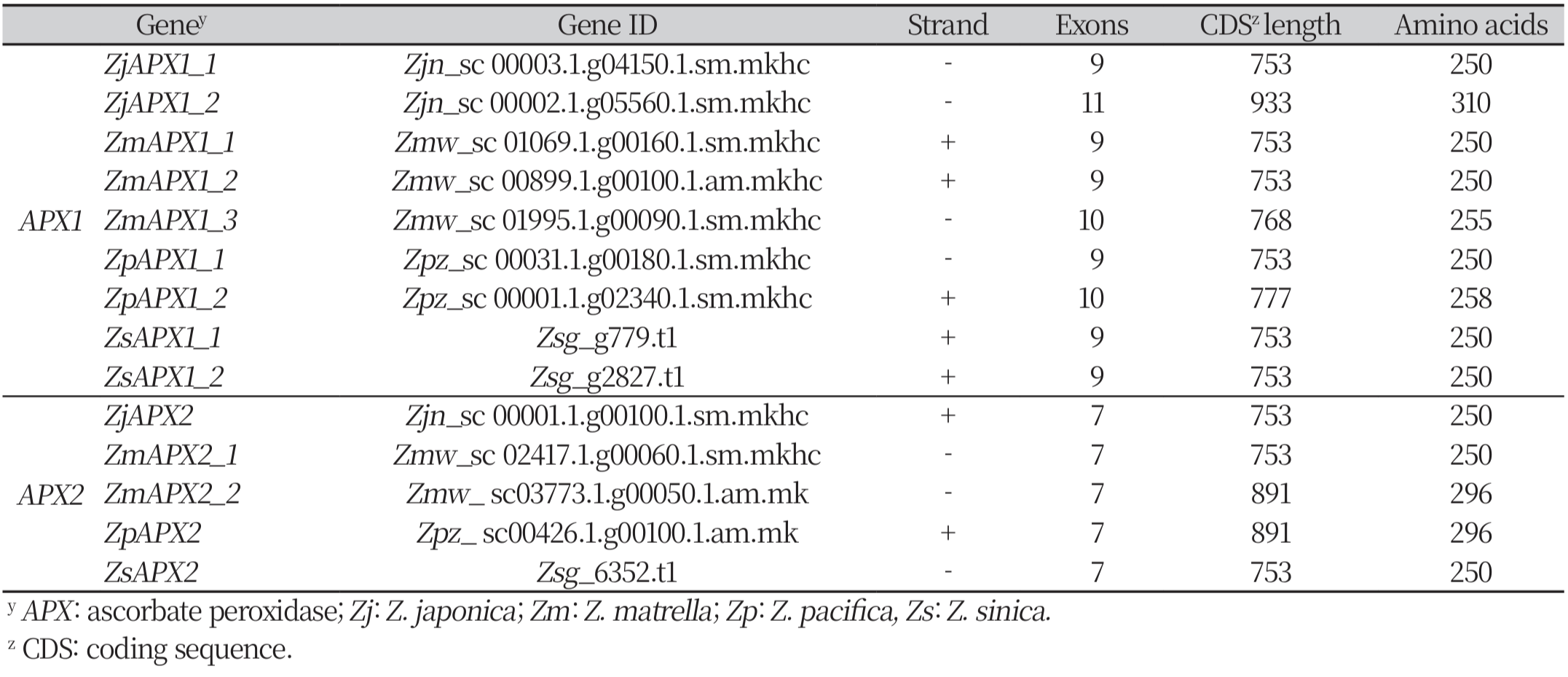
Fig. 1
Phylogenetic relationship of APX genes among the four species of zoysiagrass using maximum likelihood method with 500 bootstrap replications. The blue boxes represent the ascorbate peroxidase domain (cd00691), orange boxes represent the plant peroxidase like super family domain (cl00196), and black boxes represent exons and lines represent introns. A and B denotes major groups, and a-f denotes subgroups. APX: ascorbate peroxidase; Zj: Z. japonica; Zm: Z. matrella; Zp: Z. pacifica; Zs: Z. sinica.
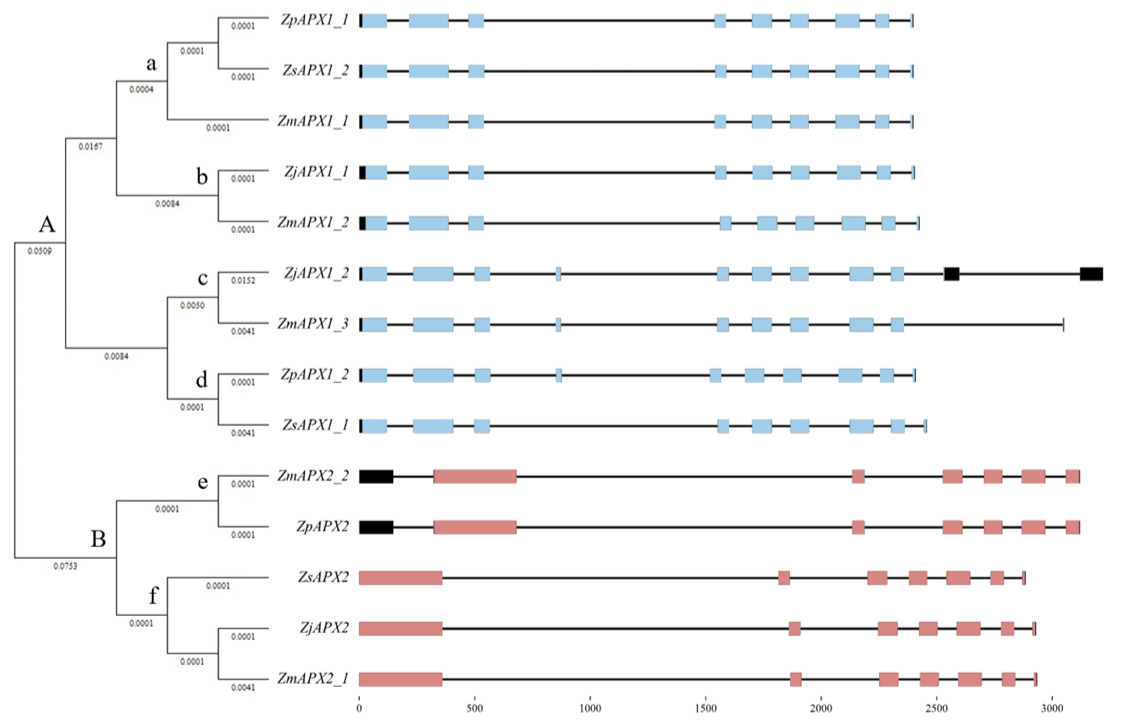
Fig. 2
Multiple sequence alignment of the predicted APX proteins in four species of zoysiagrass. The colors correspond to the amino acid identity and the bar graph indicates the degree of conservation between the sequences. The predicted domains and variants are indicated by the boxes. APX: ascorbate peroxidase; Zj: Z. japonica; Zm: Z. matrella; Zp: Z. pacifica; Zs: Z. sinica; SAP: single amino-acid polymorphism.
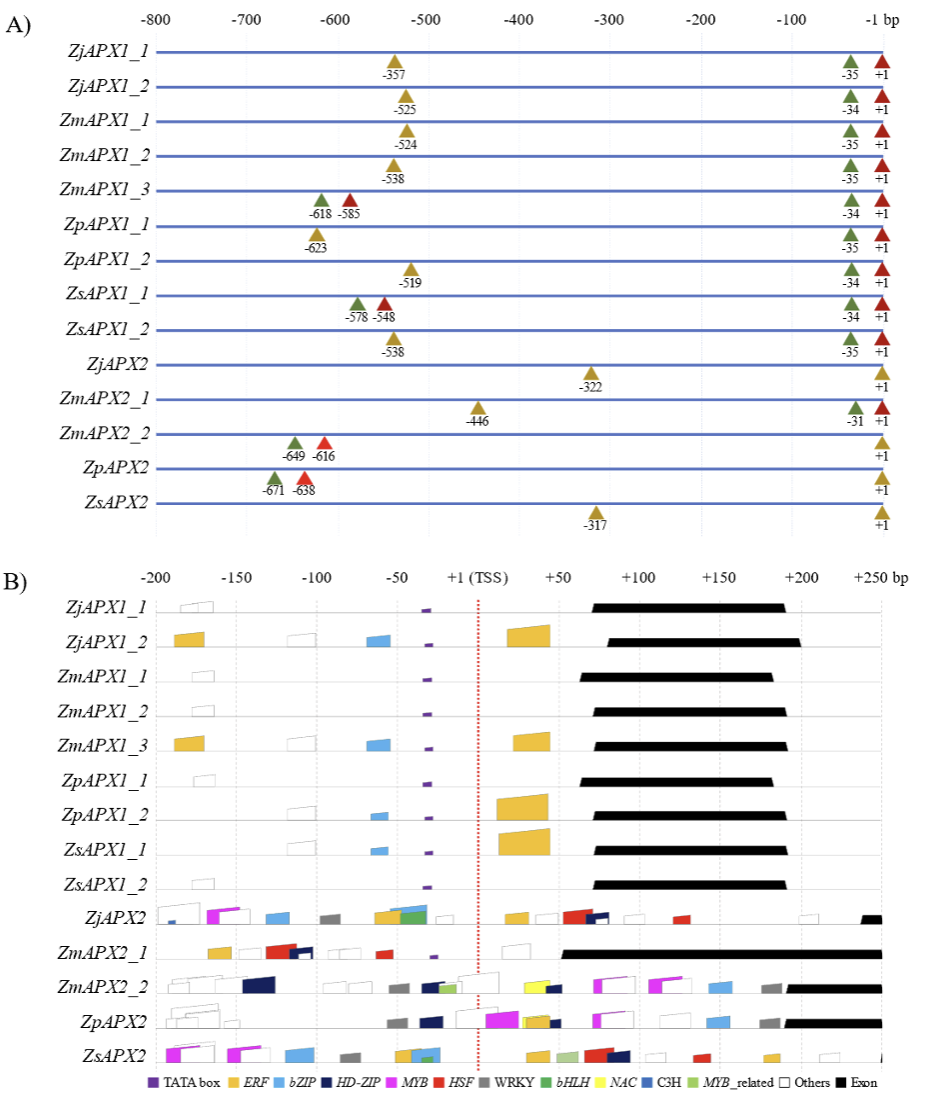
Zoysia APX upstream and downstream regulatory region analysis and identification of transcription factor binding sites
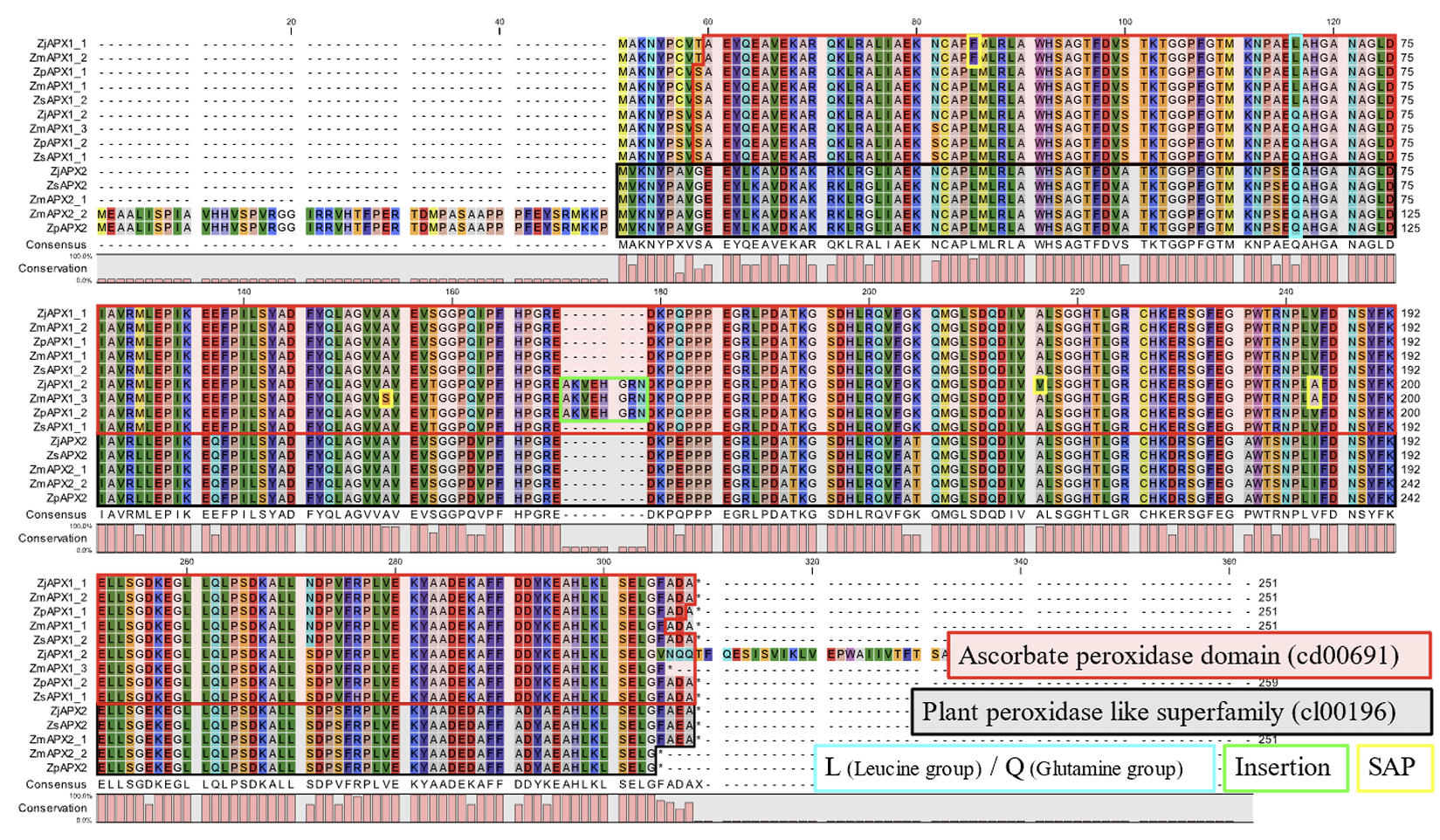
The TF binding site analysis was conducted in regions 200 bp upstream of the TSS and downstream up to the first exon. However, due to the absence of a TF database for Z. sinica, the results were derived using Z. japonica, Z. matrella, and Z. pacifica as references, and only the overlapping results were selected for analysis. As a result, 21 TF binding sites were predicted, which included ethyleneresponsive factor (ERF), basic leucine zipper (bZIP), GOLDEN2-like (G2-like), DNA-binding with one finger (Dof), homeodomain leucine zipper (HD-ZIP), v-myb avian myeloblastosis viral oncogene homolog (MYB), heat shock factor (HSF), and WRKY (Table 2; Fig. 3B). Among these, TFs such as ERF, bZIP, HD-ZIP, MYB, HSF, and WRKY are known to play a role in responses to environmental changes and stress reactions (Cao et al., 2020; Guo et al., 2021; Guo et al., 2016; Guo et al., 2024; Javed et al., 2020; Li et al., 2022; Yang et al., 2014). Notably, ERF binding sites were predicted with the highest frequency. ERF plays a critical role in the transcriptional regulation of various biological processes by binding to promoter regions. These processes include plant growth and development, as well as responses to abiotic stresses such as drought, salinity, heat, and cold. They also encompass biotic stresses like wounding (Cheng et al., 2023; Nakano et al., 2006). This suggests that APX functions as a stress-related gene, and these transcription factors are likely involved in regulating its expression.
Most TFs were predicted in the APX2 group, which aligns with the known role of TATA-less TSSs. Compared to TSSs with TATA boxes, TATA-less TSSs rely more heavily on regulators for expression control and are influenced by multiple factors (Zhou et al., 2008). Furthermore, apart from ERF and bZIP, most TFs involved in responses to environmental changes and stress were predicted in the APX2 group, suggesting that the APX2 group may contribute to the adaptation of zoysiagrass to environmental changes and stress conditions.
Table 2
List of predicted transcription factors interacting APX genes in four species of zoysiagrass.
Fig. 3
Analysis of the upstream and downstream regulatory region of the APX genes predicted in the four zoysiagrasses. A) The predicted TSS and TATA box sites are represented as triangles. Red triangles indicate TSS (TATA+), yellow triangles indicate TSS (TATA-), and green triangles represent TATA boxes. The TSS is marked as +1. The predicted TATA boxes are located too far from the gene in ZmAPX2_2 and ZpAPX2, and no TATA box was predicted for ZjAPX2 and ZsAPX2. B) TFs predicted upstream (200 bp) and downstream (up to the first exon) of the TSS are shown. Environmental and stressrelated TFs are indicated by colored boxes, while other TFs are represented with white boxes (see legend for details). APX: ascorbate peroxidase; Zj: Z. japonica; Zm: Z. matrella; Zp: Z. pacifica; Zs: Z. sinica; TSS: transcription start site; TFs: transcription factors; ERF: ethylene-responsive factor; bZIP: basic leucine zipper; HD-ZIP: homeodomain leucine zipper; MYB: v-myb avian myeloblastosis viral oncogene homolog; HSF: heat shock factor; bHLH: basic helix–loop–helix; NAC: NAM, ATAF, CUC.
Protein structure prediction and the analysis of the conserved domain
The protein structures of predicted APX1 and APX2 were modeled using Sorghum bicolor APX1 (SbAPX1, 8djr.1.A; L-ascorbate peroxidase 1) as templates, which were identified as the best matches (Zhang et al., 2023). Based on the amino acid sequence alignment analysis (Fig. 2) and a comparison with the SbAPX1 structure, one insertion was observed within the conserved domain of ZjAPX1_2, ZmAPX1_3, ZpAPX_1_2, along with four SAPs. Among these, the insertion affected the protein structure of ZjAPX1_2, ZmAPX1_3, and ZpAPX1_2. Additionally, SAP at position 85 (a substitution from leucine to phenylalanine) resulted in a deficiency of a heme-binding structure in the ZjAPX1_1 and ZmAPX1_2 proteins (Fig. 4). APX, a class I protein of the plant heme peroxidase superfamily, relies on heme to use ascorbate for ROS decomposition efficiently (Maruta et al., 2016). In the model plant Arabidopsis, it has been confirmed that a knock-out of APX1 leads to the collapse of the H2O2 scavenging system, resulting in the accumulation of H2O2 and protein oxidation, thereby demonstrating that APX1 is a critical factor in ROS metabolism (Davletova et al., 2005). It is also known to be activated under drought and high-temperature stress, as well as exposure to heavy metals in plants (Gill and Tuteja, 2010). Therefore, further studies are needed to investigate how the structural differences in APX proteins predicted through in silico analysis, particularly the predicted heme-binding structure deficiency in APX1 of Z. japonica and Z. matrella, actually affect the plants’ ability to respond to stress. And two distinct groups were observed at the 116th amino acid position: the leucine group (ZjAPX1_1, ZmAPX1_1, ZmAPX1_2, ZpAPX1_1, ZsAPX1_2) and the glutamine group (ZjAPX1_2, ZmAPX1_3, ZpAPX1_2, ZsAPX1_1, ZjAPX2, ZmAPX2_1, ZmAPX2_2, ZpAPX2, ZsAPX2). Aside from differences in sequence length, the protein structures and theoretical isoelectric point (pI) values of both groups were found to be similar (Fig. 4). This distinction appears to arise from the difference in chemical properties between hydrophobic leucine and polar glutamine. This result also corresponds with the grouping observed in the phylogenetic tree (Fig. 1), confirming consistency across both analyses.
Conclusion
In this study, we predicted a total of 14 cAPX genes from four species of zoysiagrasses that respond to environmental changes, including abiotic stresses such as drought, salinity, heat, and cold. We aimed to elucidate the genetic and structural differences by comparing the predicted protein structures generated from these genes. As a result, the genes were classified into two major groups (APX1 and APX2) and six subgroups. Structurally, we identified changes in protein structure caused by amino acid insertions, with SAP-related changes predicted to cause heme-binding structure deficiency in ZjAPX1_1 and ZmAPX1_2. These structural changes suggest that there may be differences in stress responses among the zoysiagrass species. However, as this is based on in silico analysis, further research is required to evaluate the actual response levels in the plants.
Additionally, through TF analysis, we confirmed that each gene could be regulated by environmental changes and stress. We also found differences in the number of regulators depending on the presence or absence of the TATA box. A detailed analysis of the regulatory network could further our understanding of the mechanisms behind zoysiagrass’ response to environmental changes and stress.
Due to global climate change, plants are increasingly exposed to abiotic stress, including rapid temperature increases, irregular precipitation, drought, and increased soil salinity (Ullah et al., 2021). Since zoysiagrass plays a crucial role in everyday life, including leisure activities and environmental management, continuous research on factors related to environmental changes and stress is essential for its sustainable use.
This study focused only on cAPX, but there are other APX isoforms, including sAPX and tAPX in chloroplasts, as well as mAPX (Shigeoka et al., 2002). Future studies could consider extending the research through in-silico analysis of these additional APX genes in zoysiagrass. In particular, for membrane-bound APX, it would be worthwhile to confirm their presence using green fluorescent protein (GFP) molecular markers.
Fig. 4
Analysis of the physical, chemical and structural properties of APX proteins predicted in four zoysiagrass, with comparison to SbAPX1. For each APX protein, the tertiary structure was predicted using SWISS-MODEL with the image colored by Confidenceclass based on comparison and structural alignment to SbAPX1 using as a template. APX: ascorbate peroxidase; Zj: Z. japonica; Zm: Z. matrella; Zp: Z. pacifica; Zs: Z. sinica; Sb: S. bicolor; pI: isoelectric point; GMQE: global model quality estimate; Seq: sequence; HEM: heme.

Acknowledgment
This research was funded by the New Breeding Technologies Development Program (RS-2024-00322302), Rural Development Administration, Republic of Korea.
Authors Information
Yongsam Jeon, https://orcid.org/0000-0003-1168-3823
Chetan Kaur, https://orcid.org/0000-0001-6226-7921
Young-Sun Kim, https://orcid.org/0000-0002-5645-7021
Donghwan Shim, https://orcid.org/0000-0002-0223-595X
Eun-Ji Bae, https://orcid.org/0000-0003-4597-8873
Geung-Joo Lee, https://orcid.org/0000-0002-3774-1860

